










Liquid biopsy represents a groundbreaking advancement in disease diagnostics, offering a less invasive alternative to traditional tissue biopsies, particularly for cancer.
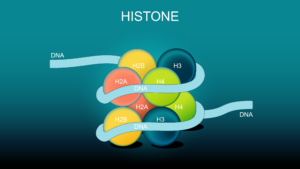
histone modifications play a crucial role in regulating gene expression and chromatin structure. These modifications work together in complex patterns to fine-tune cellular processes.
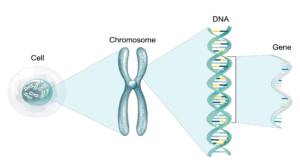
DNA methylation serves as a pivotal epigenetic mechanism that significantly influences gene regulation by recruiting proteins that facilitate gene repression or by hindering transcription factors

